�Ϻ���(gu��)�Hע��(c��)eCTD��ʽ ����(w��)���� �x���Ƽ�����(y��ng)
�l(f��)؛���c(di��n)���㽭ʡ������
�l(f��)���r(sh��)�g��2025-07-05

�l(f��)؛���c(di��n)���㽭ʡ������
�l(f��)���r(sh��)�g��2025-07-05
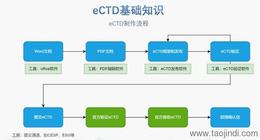
eCTD�ļ���������ѭ��(y��n)��ķ�Ҏ(gu��)Ҫ��͘�(bi��o)��(zh��n)�����̣��������P(gu��n)�IҪ�c(di��n)������eCTD����ģ�K���Y(ji��)��(g��u)������ģ�K1��������Ϣ����ģ�K5���R����(b��o)�棩���谴ICH�ͱO(ji��n)�C(j��)��(g��u)Ҫ��(g��u)��Ŀ䛘�(sh��)���w�����x���ļ��ύ�Ӽ�(j��)����***���(b��o)�r(sh��)�_�������ã�����ԭ�Ϻ��Ƅ����¹�(ji��)���硢���谴���^���w���Ȳ�֣��o�φΪ�(d��)���¡�PDF�����ӕ�(sh��)������(d��o)��Ŀ䛣��ͳ�朽ӣ���W(w��ng)�(y��)���D(zhu��n)�������^(gu��)5�(y��)���ļ���횰���Ŀ䛣�TOC/LOT/LOF�������g(sh��)����(sh��)����ʼҕ�D���O(sh��)��Ĭ�J(r��n)�s�ż�(j��)�e���(y��)�沼�֣���(sh��)��չ�_(k��i)�Ӽ�(j��)�����^(gu��)����(j��)�����ļ���С��������(b��o)ϵ�y(t��ng)���ơ��(y��n)�C���ߣ�ʹ��ܛ������BXeCTD���Ԅ�(d��ng)���ɕ�(sh��)���ͳ�朽ӣ���ͨ�^(gu��)����У�(y��n)��PDFУ�(y��n)���ܴ_����Ҏ(gu��)�ԡ� �W��CESP�ύͨ�����P(gu��n)���g(sh��)֧�֡��Ϻ���(gu��)�Hע��(c��)eCTD��ʽ
��(j��ng)��(j��)Ӱ��c�ɱ�Ч�� �M�ܳ���Ͷ���^�ߣ�ƽ��ÿ��I(y��)��50�f(w��n)�WԪ������eCTD�ɜp��30%�Č��u(p��ng)���t�ɱ����L(zh��ng)��Ч�档����ˎ��I(y��)ͨ�^(gu��)eCTD��(f��)��ԭ�Д�(sh��)��(j��)����(ji��)ʡ80%�����(b��o)��(zh��n)��r(sh��)�g���W���A(y��)��ܿ�2�|�WԪ�Y����С��I(y��)��ɔ�(sh��)�ֻ��D(zhu��n)�͡� ���팏���c��(sh��)��(j��)�[˽ eCTD�еĻ��ߔ�(sh��)��(j��)��������̎�������ϡ�ͨ�Ô�(sh��)��(j��)���o(h��)�l������GDPR��Ҫ���R��ԇ�(y��n)?z��i)��K��ģ�K5�����ύ�踽������ί�T��(hu��)����(zh��n)�ļ����҅^(q��)��汾���w�F(xi��n)����(gu��)���팏����AI�o�������������ڱ��o(h��)�[˽��ͬ�r(sh��)������(sh��)��(j��)̎��Ч�ʡ� ���g(sh��)�ں��c���I(l��ng)��(y��ng)�� eCTD��ʽ�U(ku��)չ���t(y��)����е�ͱ���Ʒ�I(l��ng)�W��ԇ�c(di��n)eCTD-MDR�(xi��ng)Ŀ����ISO��(bi��o)��(zh��n)������a(ch��n)Ʒ��eCTD�踽�����ﰲȫ��(sh��)��(j��)��(k��)�����c�W�˻����(k��)��(sh��)�r(sh��)ͬ����δ��(l��i)��eCTD���c��ӽ����n����EHR��ϵ�y(t��ng)��(du��)�ӣ�֧�ւ�(g��)�Ի���ˎ�� ���m(x��)���M(j��n)�c�ИI(y��)�����C(j��)�� EMAÿ��l(f��)��eCTD��(sh��)ʩ��(b��o)�棬������Ҋ(ji��n)�e(cu��)�`����ָ�ϡ��ИI(y��)(li��n)�ˣ���EFPIA��ͨ�^(gu��)������ӑ��(hu��)��O(ji��n)�ܙC(j��)��(g��u)�������g(sh��)ʹ�c(di��n)���Ƅ�(d��ng)��(bi��o)��(zh��n)��(y��u)�����_(k��i)��ʽAPI�ӿڵ��ƏV�����M(j��n)eCTD����朵Ļ������ԣ����ͼ��g(sh��)�i���L(f��ng)�U(xi��n)������CDE eCTD��(b��o)�r(ji��)����(gu��)ESG����ύͨ����Ո(q��ng)���P(gu��n)���g(sh��)֧�֡�
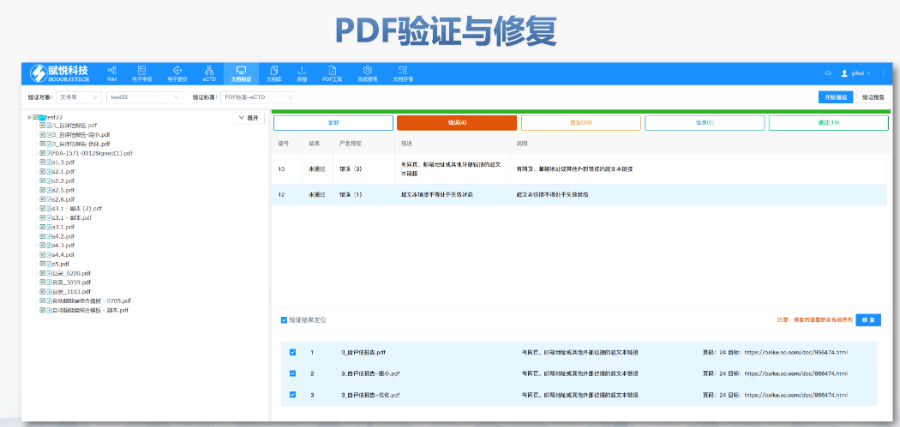
�ļ��������ڹ�����eCTD֧���ļ���Q��Replace�����h����Delete���Ȳ������������ļ������磬���R���о������r(sh��)����Replace�������w�f�汾������(xi��n)�ύ��BaselineSubmission���������a(b��)��vʷ���|(zh��)�Y�ϣ������ڷ��溯�����o(w��)��(n��i)��׃�����R����(sh��)��(j��)�c�о���(bi��o)���ļ���STF����ģ�K4��5�е��о���(sh��)��(j��)��ͨ�^(gu��)STF��StudyTaggingFiles�����ã��_����(sh��)��(j��)�c�ęn�P(gu��n)(li��n)��FDAҪ��(sh��)��(j��)������SASXPORT��ʽ��������ģ�K3-5���҆�(g��)�ļ����^(gu��)4GB���֡�2022��y(t��ng)Ӌ(j��)�@ʾ��58%��ANDA���о���(sh��)��(j��)���g(sh��)�ܽ^��(bi��o)��(zh��n)��TRC���e(cu��)�`���ܡ���Ӻ����c����Ҫ��FDA������356h��1571����ʹ�Ô�(sh��)�ֺ�����PDF�ļ���ֹ���ܻ��O(sh��)�þ����ơ���Ӻ��������21CFRPart11Ҏ(gu��)�����_�������(y��n)�C�����ɷ��J(r��n)�Ժ͔�(sh��)��(j��)�����ԡ��������(w��)�cϵ�y(t��ng)��Q�������x���Ƽ���Ӌ(j��)�ύ��2000��eCTD��Ո(q��ng)������ɽ���40%�˹��e(cu��)�`�ʡ�
���(b��o)�����cҪ�� �Y�Ϝ�(zh��n)�� ��(n��i)��Ҫ�����a(ch��n)Ʒ���������a(ch��n)��ˇ��ԭ���ρ�(l��i)Դ���O(sh��)�䅢��(sh��)�ȣ����|(zh��)�����Ƙ�(bi��o)��(zh��n)��SOP����(w��n)���Ԕ�(sh��)��(j��)������ȫ���c�����о��ȡ� ��ʽҎ(gu��)���� ����CTD��ͨ�ü��g(sh��)�ļ�����ʽ����ģ�K���¹�(ji��)����ģ�K3��CMC��(sh��)��(j��)���� ����ύ�����eCTD��(bi��o)��(zh��n)���ļ�С��10GBͨ�^(gu��)ESGϵ�y(t��ng)�ύ�����^(gu��)���x��CD-ROM���� �ύ�cע��(c��) �A(y��)����DMF̖(h��o)�������ύǰ��Ո(q��ng)���_���ļ��c��̖(h��o)������ �ڙ�(qu��n)��(sh��)��LOA������������DMF���Ƅ��S(ch��ng)���ṩ�ڙ�(qu��n)�ţ����_�ɲ�醵��¹�(ji��)�� �M(f��i)�ã����(l��i)ԭ��ˎDMF���U�{���M(f��i)��2024��s9,468��Ԫ���� FDA�������� �������u(p��ng)��2-3�܃�(n��i)�_�J(r��n)�ļ������ԡ� �����Ԍ��u(p��ng)��CA����ᘌ�(du��)���(l��i)DMF���s60�졣 ���g(sh��)���u(p��ng)����DMF���Ƅ���Ո(q��ng)����ANDA��NDA�����Õr(sh��)����(d��ng)������60-180�졣 �Y(ji��)��������FDA����Ҫ���a(b��)�䔵(sh��)��(j��)����DMF����o(w��)������(zh��n)����B(t��i)��ͨ�^(gu��)������յ����o(w��)�M(j��n)һ����Ҋ(ji��n)������No Further Comment Letter�������ô�ANDAע��(c��)���(b��o)���P(gu��n)���g(sh��)֧�֡�
�ļ��|(zh��)����ӵĚvʷ�^(gu��)�� 2017��ǰ������(gu��)���S���|(zh��)�ceCTD�����ύ�����˺�����̭���|(zh��)ͨ���������o����r�µ����⌏����2020����ӻ�������IND��NDA��ANDA��DMF��(qi��ng)�Ʋ���eCTD��ʽ�� ϵ�y(t��ng)ƽ�_(t��i)����(j��) FDAͨ�^(gu��)��ˎƷ�I(y��)��(w��)��(y��ng)��ϵ�y(t��ng)���͡�ˎƷeCTDע��(c��)ϵ�y(t��ng)����(sh��)�F(xi��n)����Y�Ͻ��ա������c���u(p��ng)��ȫ���̔�(sh��)�ֻ���2022��ϵ�y(t��ng)���Ԅ�(d��ng)���������ĕ�(sh��)�Ͷ������ѹ��ܣ��p���˹����A(y��)�� ����ęn�Y(ji��)��(g��u)��(y��u)�� ����(gu��)eCTD���÷��ļ��A�Y(ji��)��(g��u)�����绯�W(xu��)ˎƷ��ģ�K1-5�քe��(du��)��(y��ng)�����ļ������Y(ji��)��(b��o)�桢�|(zh��)����(sh��)��(j��)�ȡ�2020��������R��ԇ�(y��n)��(sh��)��(j��)��(k��)���b��Ҫ��(qi��ng)����(sh��)��(j��)�����ԡ��W��eCTDע��(c��)��ԃ(x��n)���P(gu��n)���g(sh��)֧�֡�����eCTD��ʲô
eCTD�(y��n)�C��(sh��)�`�փ�(c��)���P(gu��n)���g(sh��)֧�֡��Ϻ���(gu��)�Hע��(c��)eCTD��ʽ
eCTD�(y��n)�C��(bi��o)��(zh��n)�ć�(y��n)�����c���(l��i)���W�ˌ�(du��)eCTD���(y��n)�CҪ��֞顰�e(cu��)�`�������桱�͡���ʾ��Ϣ������(j��)�����С��e(cu��)�`���(xi��ng)ֱ�ӌ�(d��o)�����(b��o)���ܡ��(y��n)�C�(xi��ng)Ŀ���w�����(l��i)��149�l�������ļ�����Ҏ(gu��)������·���L(zh��ng)�����ƣ���PDF���x�ԣ���ֹ�ܴa���o(h��)����XML�Ǽ��ļ������Եȡ����磬�ļ��U(ku��)չ����횷���Ҏ(gu��)������.xpt�����R����(sh��)��(j��)���������ļ��A�Ӽ�(j��)������Ŀ䛻��ϴ���ļ������^���Ї�(gu��)��������(b��o)�(y��n)�C��(bi��o)��(zh��n)���ĺ�(ji��n)���棨54�l�����W�˵��(y��n)�C�wϵ�����(f��)�s���w�F(xi��n)����ߘ�(bi��o)��(zh��n)�ļ��g(sh��)�O(ji��n)�ܡ��Ϻ���(gu��)�Hע��(c��)eCTD��ʽ
������̶��Ԓ(hu��)��Ո(q��ng)?ji��n)څ^(q��)̖(h��o)�������"-"�� �(xi��)�֙C(j��)̖(h��o)�������ˈ�(b��o)�r(ji��)�����M(f��i)���ն���֪ͨ